
Contact
Email: kwa5025@umd.edu
Call: (240) 314-6285
Kyle Anderson
Assistant Research Scientist
NIST
Contact
Email: kwa5025@umd.edu
Call: (240) 314-6285
Education
- NIST-NRC Postdoctoral Research Associate, 2015-2016
- PhD, Biochemistry, University of Maryland College Park, 2015
- BS, Biochemistry and Molecular Biology, Pennsylvania State University, 2010
Profile
Kyle Anderson is the Project Leader of HDX-MS for Biopharmaceutical Analysis at NIST. In addition to using hydrogen/deuterium exchange mass spectrometry (HDX-MS) to characterize the structural dynamics of biopharmaceuticals, he is active in developing and promoting adoption of state-of-the-art methods, instrumentation, and software for HDX-MS to ensure quality and accelerate development of biotherapeutics and biosimilars. He received the U.S. Department of Commerce Silver Medal in 2021 and Bronze Medal in 2023 and the Presidential Early Career Award for Scientists and Engineers in 2022. He is a member of the Editorial Advisory Board of JASMS, the International Society of HDX-MS, the American Society for Mass Spectrometry, and the CCQM Protein Analysis Working Group and its Protein Structure and Activity Task Group.
Postdoctoral Research Opportunities
NIST-NRC Postdoctoral Research Associate Program: Rolling application deadlines are Feb 1 and Aug 1. US citizens only. $82,764 (2024) annual salary plus benefits, relocation, travel, and research funding. Contact Kyle Anderson before applying. View Job Posting.
Positions for non-US citizens: Contact Kyle Anderson directly.
CURRENT RESEARCH TOPICS
- Chromatography at -30 °C for reduced back-exchange and improved dynamic range for HDX-MS
- Characterization of protein-based vaccines in formulation by HDX-MS
- Characterization of drug-protein interactions by HDX-MS
- Adoption of internal H/D exchange reporters for standard reference materials to improve reproducibility and harmonization of HDX-MS
RECENT RESEARCH PROJECTS
Subzero chromatography system for reduced back-exchange and carryover and improved dynamic range
For hydrogen-deuterium exchange mass spectrometry (HDX-MS) to have an increased role in quality control of biopharmaceuticals, H for D back-exchange occurring during protein analyses should be minimized to promote greater reproducibility. Standard HDX-MS analysis systems that digest proteins and separate peptides at pH 2.7 and 0 °C can lose >30% of the deuterium marker within 15 min of sample injection. We developed an analysis system for HDX-MS that conducts chromatography at ‑30 °C, reducing back-exchange and enabling chromatographic gradients well beyond an hour. Chromatographic resolution is improved, allowing HDX-MS measurements on a greater number of peptides. Residual back-exchange of ≤ 10% still occurs, mostly due to preparation zone at 0 °C. The average peptide eluted during a 40 min gradient contained ≈16% more deuterium than peptides eluted with a conventional 8 min gradient at 0 °C. A subset of peptides exhibited ≈26% more deuterium. Although chromatographic peaks shift with EG concentration and temperature, the apparatus elutes unbroadened LC peaks. Electrospray ion intensity does not decline with increasing EG fraction. To minimize bias from sample carryover, the fluidic circuits allow flush and backflush cleaning of all enzyme and LC columns. The system can perform LC separations and clean enzyme columns simultaneously. Reproducibility is enhanced by regulating temperature zones to ±0.05 °C.
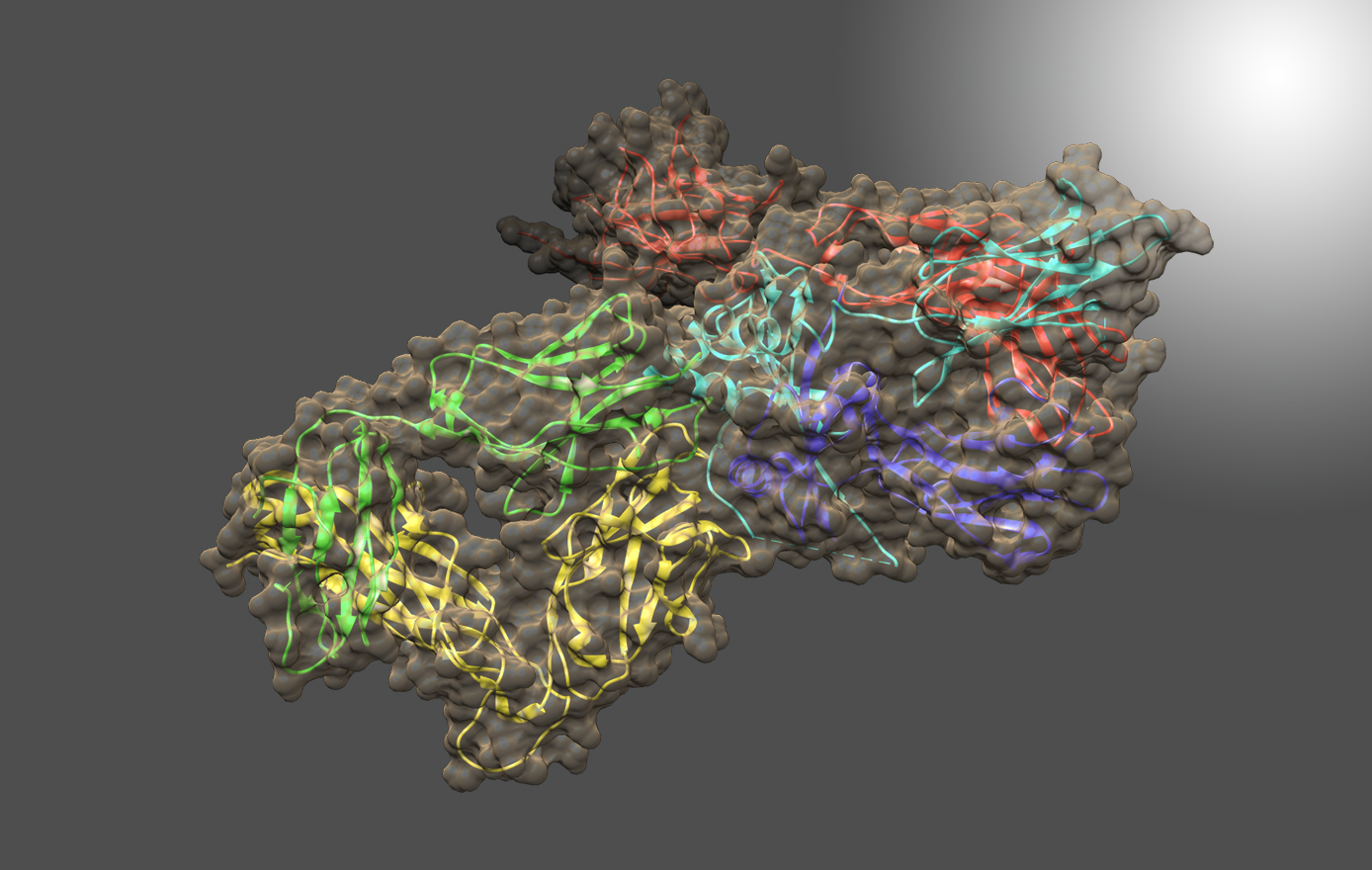
Interlaboratory comparison of HDX-MS measurements using Fab of NISTmAb
An interlaboratory study involving 37 co-authors in 15 unharmonized laboratories determined the reproducibility of continuous-labeling, bottom-up HDX-MS measurements by measuring the deuterium uptake of Fab of NISTmAb RM8671 (PDB: 5K8A) for immersions in D2O for tHDX = (30 to 14400) s and THDX = (25, 21, 3.6) °C. Each laboratory was sent a standardized kit containing Fab protein and the reagents necessary to conduct experiments. Laboratories reported ≈89,800 centroid measurements for 430 proteolytic peptide sequences of the Fab fragment (≈78,900 centroids), giving nearly 100% coverage, and approximately 10,900 centroid measurements for 77 peptide sequences of the Fc fragment. Nearly half of peptide sequences are unique to the reporting laboratory, and only two sequences are reported by all laboratories. The majority of the laboratories (87%) exhibited centroid mass laboratory repeatability precisions of ≤ (0.15 ± 0.01) Da. All laboratories achieved ≤ 0.4 Da. For immersions of protein at THDX = (3.6 to 25) °C and for D2O exchange times of tHDX = (30 s to 4 h) the reproducibility of back-exchange corrected, deuterium uptake measurements for the 15 laboratories is (9.0 ± 0.9)%. A 9 lab cohort that immersed samples at THDX = 25 °C exhibited reproducibility of (6.5 ± 0.6)% for back-exchange corrected, deuterium uptake measurements.
Effects of IgG1 glycoforms on interactions with receptor FcγRIa (CD64a)
HDX-MS and molecular dynamics (MD) simulations were used to study stabilization of the IgG1-FcγRIa (CD64a) complex, particularly via intermolecular glycoprotein bonds. Discord in literature models for stabilization of the Fc-FcγRI complex and lack of data on stabilizing glycoprotein contacts between glycans of IgG1 and a conserved FG-loop (MGKHRY) of FcγRIa prompted investigation. IgG1s containing glycans with N-acetylglucosamine, galactose, and α2,6-N-acetylneuraminic terminations were measured in addition to their complexes with the FcγRIa receptor using HDX-MS and classified for dissimilarity with Welch's ANOVA and Games-Howell post hoc procedures. Additionally, they were modeled by MD simulations. For each glycoform of IgG1-FcγRIa, peptides of Fab, Fc and FcγRIa reported distinct HDX rates. MD simulations supported the differences discovered by HDX-MS through calculation of the time that transient glycan-peptide bonds exist. Results showed that stability of IgG1-FcγRIa complexes correlate with the glycoprotein interactions between IgG1 glycans and the FG-loop of FcγRIa. Additionally, intramolecular glycan-protein bonds stabilize the Fc in both isolated and complexed IgG1.
Automated removal of phospholipids from membrane proteins
A new, automated approach was developed to study structural dynamics of membrane proteins, the most common targets for pharmaceuticals. The new method involves isolation of proteins from phospholipid bilayers using a scheme that is compatible with robotic handling. The key development was the introduction of ZrO2 microbeads, which strongly coordinate with the phospholipids, and then removal of microbead-coordinated phospholipids by inline filtration. Protein is thus freed from phospholipids, which interfere with analysis by LC-MS. Previous HDX-MS publications on membrane protein dynamics removed phospholipids manually, which reduced reproduciblity and increased analysis time. The new approach was demonstrated on the transmembrane protein FcγRIIa, and showed information about the regions outside of, across, and inside of its liposomal membrane, enabling measurement of structural dynamics of the whole protein. This approach is the first to enable fully automated removal of phospholipids for HDX-MS on full-length transmembrane proteins in lipid bilayers.